
Our Research
Complete genome sequencing projects have generated enormous amounts of data. Although progress has been rapid, a significant proportion of the genes in any given genome are either unannotated or possess a poorly characterised function. Our group aims to detect, predict and describe the functions of genes, proteins, mRNAs and regulatory non-coding RNAs as well as their interactions in living organisms and their implications for disease. We are a multi-disciplinary team using wet-lab experimental techniques, computational biology and high-throughput genomics to solve these problems. Our work encompasses a number of areas:
The epitranscriptome - RNA modification and methylation
Gene regulation is a fundamental process that underpins cell development, differentiation, function and homeostasis. The discovery of epigenetic marks on both DNA and histones heralded a new era in our understanding of gene regulation. We believe that we are on the cusp of another wave of critical biological discoveries regarding similar marks placed on mRNAs and regulatory RNAs. These events sculpt the transcriptome altering how mRNAs are processed, spliced, translated, degraded and how they interact with proteins and other RNAs. Understanding this fundamental process is of critical importance to further our understanding of biological regulation and understanding mechanisms that underlie disease aetiology and susceptibility.
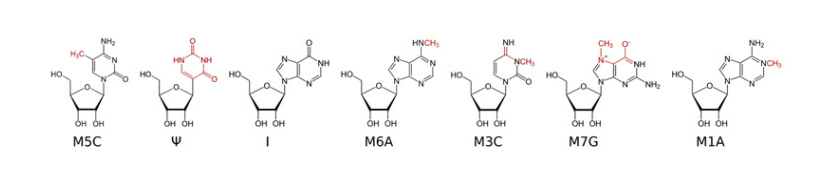
Methylated Nucleosides in RNA.

Student sequencing RNA directly using ONT MinION device in our lab
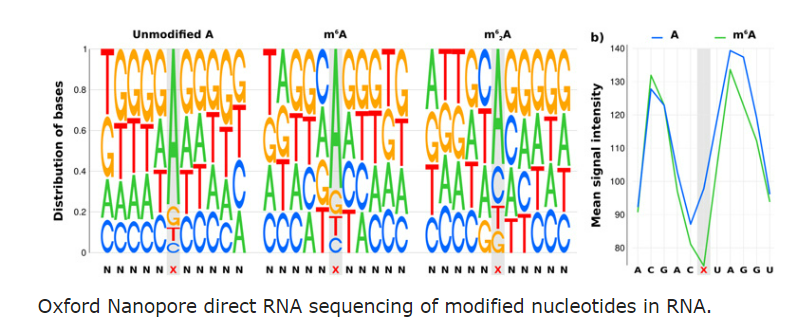
To fully explore this growing world of epitranscriptomic modifications and their effects, we are combining cutting-edge genomics techniques including Illumina high-throughput sequencing, Oxford Nanopore Direct RNA sequencing and single nucleotide mass-spectrometry to try and detect RNA modifications. Over the coming years we will be developing novel experimental protocols, algorithms and methods for detecting and describing RNA modifications and their impact in living systems.
non-coding RNA research
We have been working for over 20 years to understand how small RNAs such as microRNAs (miRNAs) regulate transcription and mRNA stability. We developed one of the first algorithms for miRNA target detection (miRanda) and worked with miRBase to make the first computational miRNA targets available to the community. The advent of next-generation sequencing made the detection of miRNAs and their global, transcriptome-wide effects, far easier to detect and allowed us to work directly in experimental systems where we could directly measure the effects of microRNAs in model organisms and human disease. We also developed a range of algorithms and web-based analysis platforms to aid the community involved in this field.
Additionally, we worked extensively on other classes of non-coding RNA molecules such as piwi-RNAs and long non-coding RNAs. Recently, we explored how processing of small RNA sequencing datasets allows the detection of 3' modification events such as uridylation and Adenosine to Inosine RNA editing.
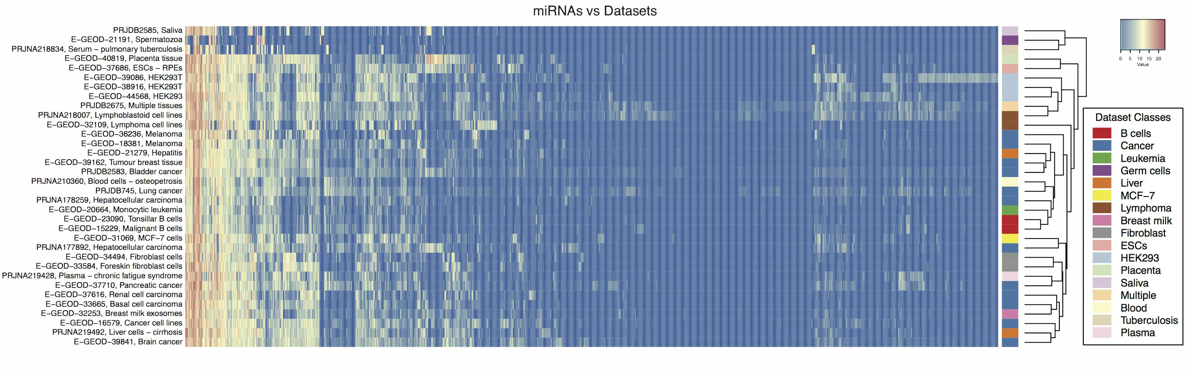
Modification profiles for microRNAs present in human cancer datasets detected using ChimiRa.
Elucidating the functions of non-coding RNAs in biological processes and disease
We have worked on the roles of miRNAs and lncRNAs in a variety of biological systems and disease models. Our algorithm Sylamer has been widely used to assess the effects of miRNAs on transcriptome level data obtained from sequencing in model organisms, cell lines and patient samples. This has allowed us to establish roles for a number of miRNAs with our colleagues and collaborating laboratories including: Maternal-Zygotic transition, Mouse models of deafness, Ichtyosis and a range of cancers. We work closely with Profs. Matthew Murray and Nicholas Coleman on the roles of miRNAs in paediatric germ cell tumours and on HPV pathogenesis.
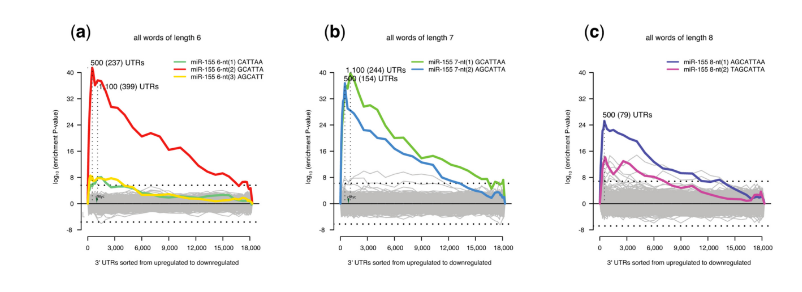
Sylamer motif enrichment profiles showing microRNA motifs present in T-helper cells from mice deficient in miR-155
Machine Learning and Data Visualisation in biology
We have an ongoing interest in the analysis of large datasets using machine learning techniques such as SVMs, RVMs, unsupervised clustering (e.g. Markov Clustering), Random Forests and Logistic Regression. Recently, we released miRNovo, a machine learning microRNA classifier for accurate prediction of miRNAs from large-scale sequencing projects. We have developed a number of tools for large-scale visualisation of biological networks including the BioLayout tool developed with BBSRC funding in collaboration with Prof. Tom Freeman (University of Edinburgh).
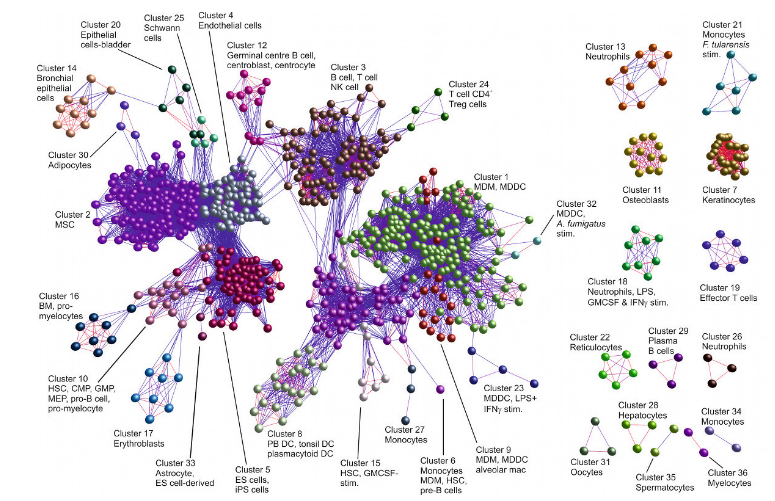
BioLayout Express 3D visualisation of samples from a human cell atlas (Tom Freeman) based on Markov Clustering (MCL)
We welcome visitors to the laboratory who wish to undertake their own experimental, computational or combined analyses under our guidance. We are always happy to talk to prospective students, visitors and lab members.

Prof. Anton J. Enright
Professor of RNA Biology and Genomics and Principal Investigator
|
Dr Carole Sargent Senior Research Associate |
Stephanie Wenlock Computational Biologist & Ph.D. Student |
Dr Julien Bauer Computational Biologist |
|
Dr Alexandra Karcanias Research Associate |
Christopher Reitter Chief Research |
Marta Andrada Almeida Chief Research |
|
Kerry Harvey Chief Research |
Alessandra Learmount PhD Student |
Melanie Maranian PhD Student |
|
Matt Morgan PhD Student |
Dr. Tatyana Lapa-Enright Visiting Researcher - Gut Brain Skin Axis |









Lab Alumni
Staff Scientists
- Carole Sargent - Cambridge University
- Stijn van Dongen - EMBL Staff Scientist
- Harpreet Saini - GSK Fellow
Postdoctoral Fellows
- Claire Quilter - Cambridge Postdoctoral Research Associate
- Adrien Leger - EIPOD Fellow
- Giovanni Bussotti - EIPOD Fellow
- Matloob Qureshi - BBSRC Fellow
- Iain Wallace - Marie Curie Fellow
- Cei Abreu-Goodger - Wellcome Trust Postdoctoral Fellow
- Russell Grocock - Wellcome Trust Postdoctoral Fellow
- Pierre Maziére- Wellcome Trust Postdoctoral Fellow
Ph.D. Candidates
- Sethlina Aryee - Cambridge Ph.D.
- Albert Kang - Cambridge Ph.D.
- Jack Monahan - Cambridge Ph.D.
- Dimitrios Vitsios - Cambridge Ph.D.
- Tommasso Leonardi - Cambridge Ph.D.
- Steve Witte - NIH Oxford Cambridge Scholars Program.
- José Afonso Guerra-Assunção - Cambridge Ph.D.
- Nenad Bartonicek - Cambridge Ph.D.
- Sergei Manakov - Cambridge Ph.D.
- Matthew Davis - Cambridge Ph.D.
M.Phil Candidates
- Sam Carling - MPhil Cambridge
- Keaan Amin - MPhil Cambridge
- Rashmi Tripathi - MPhil Cambridge
Placement & Summer Students
- Hatice Hazal Okur - Summer Student - University of Bolzano
- Dalia Bornstein - Part II Pathology Student
- Alexandra Ioana Ilie - Amgen Scholar - Polytechnic University of Bucharest
- Arinjoy Bannerjee - Part II Pathology Student
- Kate Zielinski - Part II Pathology Student
- Kelly Cheng - Part II Pathology Student
- Levia Yee - Cambridge University
- Ana Sienko - Part II Pathology Student
- Kate Lee - Part II Pathology Student
- Adam Agbamu - Cambridge University
- Magdalena Szybka - Part II Pathology Student
- Isabel Neale - Part II Pathology Student
- Tara Morrisson - Part II Pathology Student
- Florence Enjalbert - QMUL London - Orchard Therapeutics
- Swati Dahariya - British Council Exchange Student - University of Hyderabad, India
- Elsa Kentepozidou - Wageningen University, Netherlands
- Atanas Kamburov - Freie Universität Berlin
- Pedro Furio-Tari - Centro de Investigación Príncipe Felipe (CIPF), Spain